| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website https://jem.elmerpub.com |
Case Report
Volume 000, Number 000, January 2026, pages 000-000

Coexistence of Familial Acromegaly and Genetic Diabetes Mellitus Linked to Mitochondrial Dysfunction
Pavel Stancheva, f, Ekaterina Babadzhanovaa, Boris Tilovb, Maria Kraevac, Hristo Ivanovd, Meri Hristamyane
aDepartment of Endocrinology and Metabolic Diseases, Medical University of Plovdiv, Plovdiv, Bulgaria
bMedical College, Medical University of Plovdiv, Plovdiv, Bulgaria
cDepartment of Otorhinolaryngology, Medical University of Plovdiv, Plovdiv, Bulgaria
dDepartment of Medical Genetics, St. George University Hospital, Medical University of Plovdiv, Plovdiv, Bulgaria
eDepartment of Epidemiology and Disaster Medicine, Faculty of Public Health, Medical University of Plovdiv, Plovdiv, Bulgaria
fCorresponding Author: Pavel Stanchev, Department of Endocrinology and Metabolic Diseases, Medical University of Plovdiv, Plovdiv 4000, Bulgaria
Manuscript submitted August 21, 2025, accepted October 17, 2025, published online January 4, 2026
Short title: Coexistence of Familial Acromegaly and Genetic DM
doi: https://doi.org/10.14740/jem1557
| Abstract | ▴Top |
Acromegaly is a rare chronic endocrine disorder caused by excessive secretion of growth hormone (GH), usually from a pituitary adenoma, leading to characteristic somatic changes and multiple systemic complications. A substantial proportion of patients with acromegaly develop endocrine comorbidities, most notably impaired glucose metabolism and diabetes mellitus, due to GH-induced insulin resistance and β-cell dysfunction. We present a clinical case of a 37-year-old male, referred for diagnostic clarification in a specialized endocrine clinic due to an enlarged thyroid gland and distinct acromegaloid features, prompting further investigation. Biochemical testing confirmed the presence of hypersomatotropism, and magnetic resonance imaging (MRI) identified a pituitary macroadenoma, measuring 21 × 13 mm. The patient underwent successful transsphenoidal biportal adenomectomy. Given the positive family history - a sister previously diagnosed with acromegaly, genetic testing was initiated. The patient tested negative for common syndromic causes, including familial isolated pituitary adenomas (FIPA), multiple endocrine neoplasia type 1 and 4 (MEN1, MEN4). Whole exome sequencing revealed pathogenic variants in genes MT-ND5 and MT-ND4, associated with a genetic form of diabetes mellitus. While hyperglycemia may resolve in some patients after successful treatment of acromegaly, persistent diabetes mellitus was observed in our case, likely due to the alterations in MT-ND5 and MT-ND4 genes. This clinical case provides initial evidence suggesting a novel link between familial forms of acromegaly and mitochondrial diabetes, offering new perspectives on shared genetic pathways.
Keywords: Acromegaly; Macrosomatotropinoma; Familial form; Mitochondrial diabetes; MT-ND4 gene; MT-ND5 gene
| Introduction | ▴Top |
Acromegaly is a rare disease caused by hypersecretion of growth hormone (GH). Most cases of acromegaly are caused by benign microadenomas or macroadenomas of the pituitary gland, which are characterized by significant morbidity and mortality [1]. The worldwide prevalence of the disease is 2.8 - 13.7 cases per 100,000, and the incidence is 0.2 - 1.1 cases per 100,000 per year [2]. In the USA, annual prevalence estimates are higher, with around 7.1 - 8.8 per 100,000, and a European study reports a prevalence of 8.5 per 100,000 [3-5]. Incidence rates in the USA also show higher rates (0.96 - 1.17 per 100,000), while in Europe the estimates are lower at around 0.3 - 0.4 per 100,000 [3-5].
Acromegaly affects both sexes equally, with a median age of diagnosis typically in the fifth decade of life (40 - 50 years), and delays of approximately 5 years in diagnosis are common [2].
Recent data indicate an upward trend in incidence, prevalence, and age at diagnosis. In addition, the clinical presentation tends to be milder, which may affect earlier or incidental diagnosis [6, 7]. Later diagnosis results in larger tumors and the need for more complex treatment [8]. In addition, acromegaly is associated with comorbidities such as hypertension and type 2 diabetes mellitus, contributing to increased morbidity and mortality [9-11].
Although most somatotropinomas are sporadic, a small number (about 5%) have a familial pattern. More than half of the familial forms are due to multiple endocrine neoplasia type 1 (MEN-1), multiple endocrine neoplasia type 4 (MEN-4), or Carney complex [12]. In the late 1990s, several cases of familial forms of acromegaly unrelated to the syndromes mentioned above (familial isolated pituitary adenomas (FIPA)) were described, characterized by larger pituitary adenoma size and earlier diagnosis [12]. The main symptoms of acromegaly include acral overgrowth (enlarged facial features, hands, and feet) with common endocrine complications, such as insulin resistance and diabetes mellitus, hypopituitarism due to the compression of the normal pituitary gland, hypogonadism, thyroid gland enlargement and nodules.
We present a rare clinical case of a man with familial acromegaly, diagnosed on the basis of a macrosomatotropinoma. A notable aspect of this case is the co-occurrence of persistent diabetes mellitus despite successful tumor resection, which was ultimately linked to pathogenic mitochondrial DNA variants (MT-ND5 and MT-ND4). This case highlights a potential novel association between familial acromegaly and mitochondrial diabetes. To our knowledge, this is the first reported case of familial acromegaly with confirmed mitochondrial variants.
| Case Report | ▴Top |
Investigations
A 37-year-old male patient was referred to the Clinic of Endocrinology and Metabolic Diseases after an outpatient consultation with an endocrinologist due to an enlarged thyroid gland and characteristic clinical signs of acromegaly. As comorbidity, the patient presented with diabetes mellitus type 2, managed with metformin at a dose of 850 mg/day as a tolerated starting regimen. There was a relevant family history: the patient’s older sister was diagnosed with acromegaly and experienced exitus letalis due to postoperative complications following transsphenoidal adenomectomy. As a limitation, it was not possible to perform genetic testing on the patient’s sister, as no biological material was available, and such testing had not been conducted previously.
Diagnosis
Physical examination showed dark and sweaty skin. The patient had a hypersthenic habitus. The height was 180 cm, weight 105 kg, body mass index (BMI) 32.4 kg/m2. There was no evidence of gynecomastia. The feet and wrists were coarse and enlarged. Macroglossia without tooth imprints, enlarged nose and lips were observed. The thyroid gland presented enlarged with a dense-elastic consistency.
The main clinical signs of the disease in the patient are shown in Figure 1.
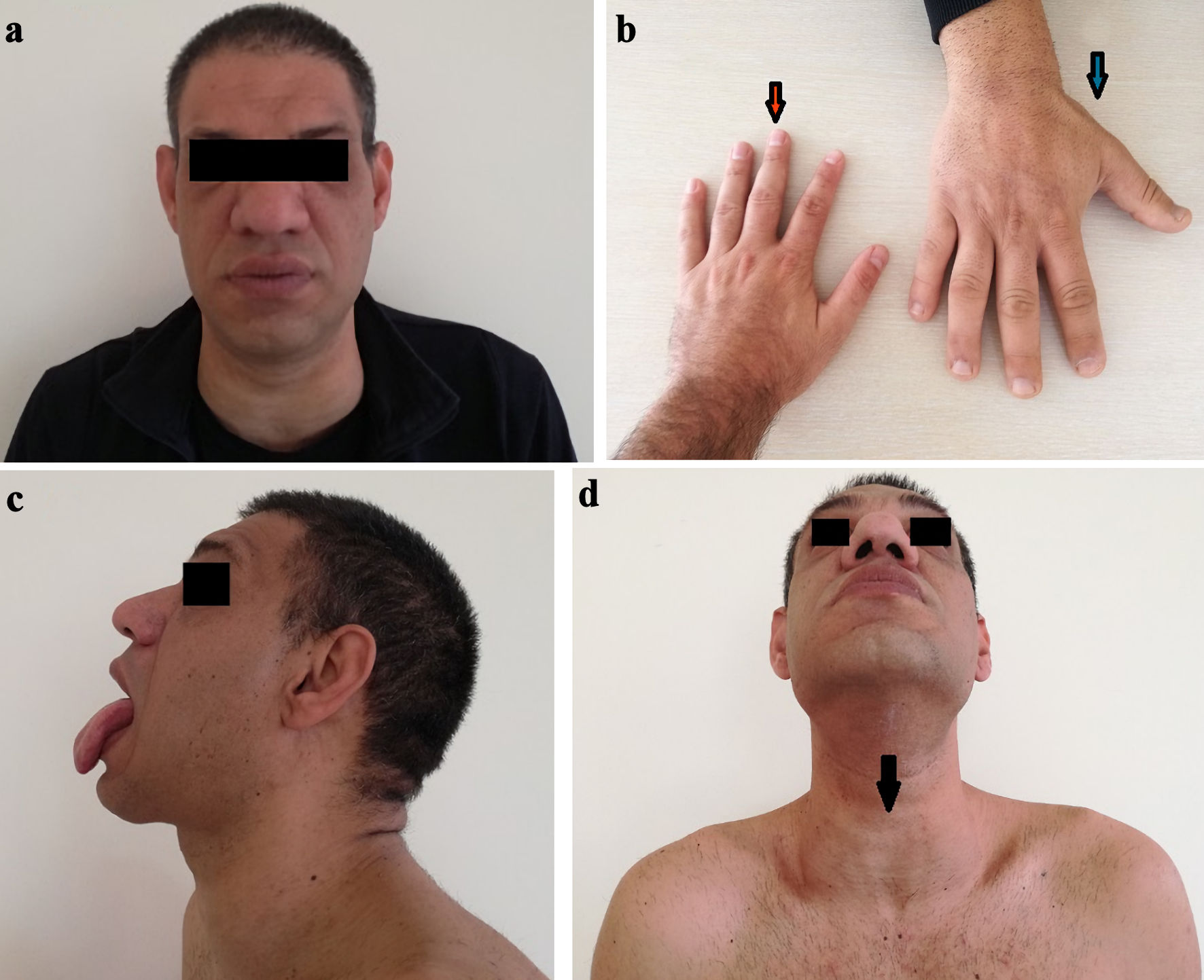 Click for large image | Figure 1. Characteristic clinical signs of acromegaly in the patient. (a) Enlarged nose and lips. (b) Enlarged and coarse wrist (blue arrow) compared to a normal wrist (red arrow). (c) Macroglossia. (d) Enlarged thyroid gland. |
The performed basic laboratory tests (Table 1) showed data on normal blood count, ionogram, and renal function. Glycated hemoglobin levels were elevated on metformin therapy. Increased GH and insulin-like growth factor 1 (IGF-1) were found, with euthyroid function, and preserved hypothalamo-pituitary-gonadal axis (luteinizing hormone (LH), follicle-stimulating hormone (FSH), testosterone, and prolactin). Thyroid autoantibodies were not elevated. No data on disorders in calcium-phosphorus metabolism were found, normal parathyroid hormone (PTH), total calcium, ionized calcium, and phosphorus were observed.
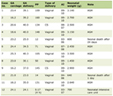 Click to view | Table 1. Basic Investigations |
Magnetic resonance imaging (MRI) of the brain was performed with a focus on the hypothalamic-pituitary region in three planes, pre- and post-contrast examination with gadolinium enhancement. An advanced extra-axial, supratentorial focal lesion with markedly expansive growth and maximum dimensions of 21 × 13 mm, localized intra-, supra-, and parasellar bilaterally, with a predominance to the left was found. The formation presented with a complex structure and relatively homogeneous intralesional characteristic - hypointensity in T1. The described pathological “mass” was irregularly rounded, with smooth and sharp contours, and in its development demonstrated aggressive biological behavior. It exteriorized suprasellarly, compressing the optic chiasm, optic nerves and tracts, and chiasmal cistern. Laterally to the left, it involved the walls of the cavernous sinus and reached the walls of the carotid siphon. After application of contrast medium (CM), it retained its hypointensity. Occlusive ventriculomegaly was absent. Focal lesions in the brain parenchyma were not visualized, nor were intra-axial tumor formations supra- or infratentorially. Ventricular system, basal cisterns, and subarachnoid spaces along the convexity were normal for age. The finding corresponded to a pituitary macroadenoma (Fig. 2).
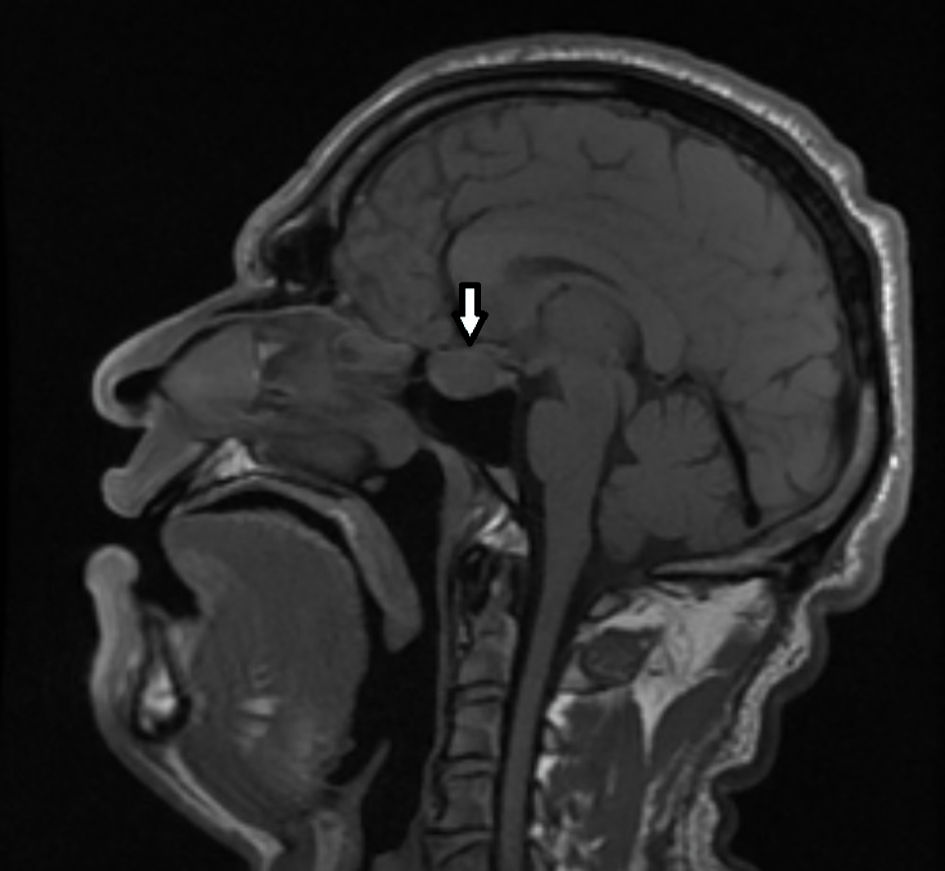 Click for large image | Figure 2. MRI of the brain showing tumor process in the sella turcica with signs of pituitary macroadenoma (sagittal plane). The arrow indicates the tumor mass. MRI: magnetic resonance imaging. |
Computerized perimetry was performed, with no evidence of visual field abnormalities. Hand radiography revealed thickened interphalangeal spaces of the distal and middle phalanges of the fingers of both hands and thickened soft tissues (Fig. 3).
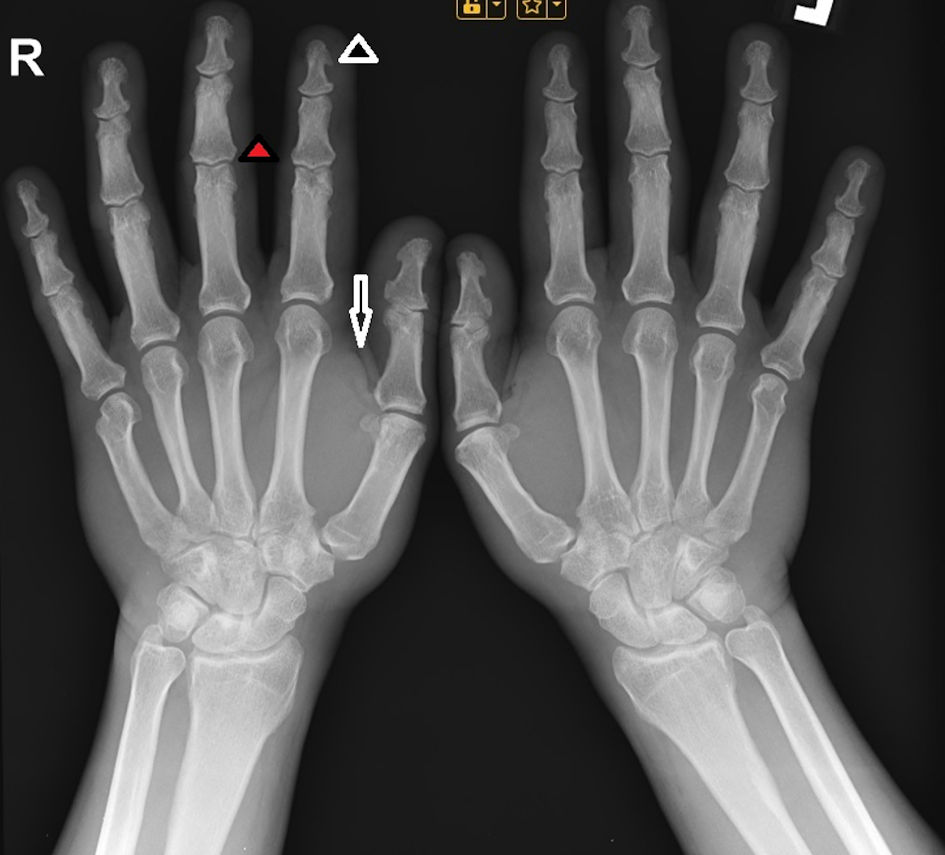 Click for large image | Figure 3. X-ray image of the hand, showing hypertrophy of the terminal tufts of the distal phalanges, forming projections similar to spurs (black arrowhead), as well as enlargement of the bases of those phalanges (red arrowhead) and thickened soft tissues (black arrow). |
The performed thyroid ultrasound visualized an enlarged nodular thyroid gland, with a total volume of 36 mL (increased for a male patient), containing multiple nodules up to 1 cm in diameter, all exhibiting benign characteristics. There were no data for enlarged cervical lymph nodes.
After consultation with a neurosurgeon, the patient was referred for surgical treatment. The tumor was extirpated using an endonasal transsphenoidal biportal approach. Pathology confirmed a sparsely granulated somatotroph adenoma with GH positivity.
Follow-up and outcomes
There were no postoperative complications. The clinical symptoms regressed, and during follow-up the patient presented with normal levels of somatotropic hormone and IGF-1. However, the patient’s concomitant diabetes mellitus persisted during follow-up. Regarding the thyroid gland, about 1 year after the surgical intervention on the pituitary gland, compression symptoms developed and a total thyroidectomy was performed. Histological examination revealed no evidence of malignant thyroid nodules, and replacement therapy with levothyroxine was started. Diabetes was managed with metformin and lifestyle modification. Follow-up studies showed improved glycemic control, with a glycated hemoglobin level of 5.37% on the same dosage of metformin; however, carbohydrate disorders persisted (glucose levels at 120 min: 8.27 mmol/L during the performed oral glucose tolerance test). Simultaneous assessment of GH levels during test showed remission (Table 2).
Click to view | Table 2. Oral Glucose Tolerance Test With a Simultanious Assessment of Growth Hormone Levels, Showing Persistent Carbohydrate Disorders and Remission of Acromegaly |
Given the presence of a family history in the patient (an older sister with acromegaly), genetic studies were conducted. Exome sequencing was performed, with analysis of the genes aryl hydrocarbon receptor-interacting protein (AIP), cyclin-dependent kinase inhibitor 1B (CDKN1B), as well as the gene encoding the menin protein. No pathogenic or likely pathogenic variants were identified to support a hereditary predisposition to the development of acromegaly. Based on the genetic analysis in the patient, FIPA, MEN1, and MEN4 were excluded.
| Discussion | ▴Top |
We presented a rare clinical case of familial acromegaly in which initial genetic analysis did not identify pathological variants associated with FIPA, MEN1, or MEN4. Subsequent consultation with a geneticist and whole exome sequencing (WES) of the patient identified candidate genes suggesting a defect in mitochondrial DNA and a potential genetic basis for the accompanying diabetes mellitus.
MEN-1 syndrome is caused by a mutation in a tumor suppressor gene located in chromosome region 11q13, encoding the protein menin, which controls cell growth, proliferation, and differentiation. Mutations in this gene can trigger the development of tumors in certain endocrine organs [13]. MEN-4 syndrome was first described in 2006 and phenotypically resembles MEN-1 - manifesting with pituitary tumors, primary hyperparathyroidism, and pancreatic tumors - but the genetic defect is in the CDKN1B gene [14].
The patient described by us phenotypically exhibits only acromegaly; there is no evidence of disorders in calcium-phosphorus metabolism due to primary hyperparathyroidism, nor are there pancreatic tumors. Genetic analysis does not confirm pathological changes in the genes responsible for the manifestation of MEN-1 and MEN-4.
In our patient, the presence of FIPA was considered, but no mutation in the AIP gene was identified. Mutations in AIP are found in only about 15-25% of described cases of FIPA [15]. In our patient’s family, his parents and children had no clinical signs of acromegaly, and no hormonal or genetic studies had been performed. A sporadic occurrence of acromegaly in both our patient and his sister has also been discussed, which is very unlikely given the frequency of acromegaly in the population. The mother of the patient had a history of diabetes.
Malicka et al describe two sisters, aged 55 and 61 years, diagnosed with acromegaly. The younger sister was treated with somatostatin analogues, with a good effect on symptoms and normalization of GH and IGF-1 levels. The older sister was treated with somatostatin, and transsphenoidal adenomectomy was performed twice due to recurrence. Genetic analysis was performed in both sisters, and genetic disorders in the direction of MEN-1 and MEN-4 were not found, as in our patient. There is no evidence whether the sisters were tested for mutations in the AIP gene [16].
WES in a patient, presenting with both diabetes mellitus and acromegaly, revealed a complex set of heterozygous variants with potential relevance to the observed endocrine phenotype. Notably, two variants were identified in mitochondrial genes associated with oxidative phosphorylation - MT-ND5 (p.F124L) and MT-ND4 (p.E87*) - both of which may impair complex I function and mitochondrial adenosine triphosphate (ATP) production, a mechanism previously implicated in mitochondrial diabetes mellitus [17]. Additionally, a frameshift variant in COL6A6 (p.D157Tfs*3), though primarily associated with muscular and connective tissue disorders, could influence pancreatic extracellular matrix integrity, potentially contributing to β-cell dysfunction [18]. Furthermore, a deleterious frameshift in SLC10A1 (p.T203Sfs*74), encoding the hepatic bile acid transporter NTCP, has been associated with altered lipid metabolism and hepatic insulin resistance [19]. Variants in MYD88 (p.E53del) and IL17RA (p.R263*) may reflect immune dysfunction, which can influence both pancreatic inflammation and pituitary regulation [20]. This may also be linked to mitochondrial oxidative stress affecting pituitary cell growth.
Although no solitary variant singularly accounts for the co-occurrence of diabetes and acromegaly, the cumulative burden of rare, potentially deleterious mutations across mitochondrial, metabolic, extracellular matrix, transport, and immune pathways may exert a synergistic effect, tipping the balance toward a complex, multi-organ endocrine phenotype. Future mitochondrial respiration assays in patient-derived cells may help confirm these findings.
In our patient, at the time of diagnosis of acromegaly, concomitant type 2 diabetes mellitus was observed as symptomatic due to high levels of somatotropic hormone. The persistence of diabetes after remission of acromegaly, along with the data obtained from WES, points to a genetic form of diabetes mellitus, specifically, a defect in β-cell function resulting from a mutation in the genes responsible for mitochondrial DNA function.
Conclusions
We present a rare case of familial acromegaly with negative genetic analysis for FIPA, MEN1, and MEN4. WES sequencing identified mutations suggestive of a possible genetic basis for diabetes mellitus due to impaired mitochondrial DNA function. Our literature search did not identify any previously described similar cases. This may also be linked to mitochondrial oxidative stress affecting pituitary cell growth, with the variants likely impairing ATP-dependent insulin secretion. Currently, there is a lack of direct studies comparing the prevalence of MT-ND4 and MT-ND5 variants in acromegalic versus non-acromegalic populations. Nevertheless, the metabolic and mitochondrial dysfunctions associated with these variants warrant further investigation to explore their potential role in endocrine disorders such as acromegaly.
Learning points
In cases of familial acromegaly with persistent diabetes mellitus, despite the successful treatment of the process causing hypersomatotropism, a potential mitochondrial etiology should be considered. Further research is needed to clarify the possible link between acromegaly and mitochondrial diabetes.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
All appropriate consent forms were obtained from the patient for the publication of the case report. Consent was also given for images and other clinical information to be reported in the journal.
Author Contributions
Pavel Stanchev: concept, literature search, data acquisition, and manuscript preparation. Ekaterina Babadzhanova: design, literature search, manuscript editing, and manuscript review. Boris Tilov: drafting of the manuscript and manuscript editing. Maria Kraeva: supervision and manuscript review. Meri Hristamyan: design, manuscript editing, translation. Hristo Ivanov: literature search, manuscript editing. All authors have reviewed the final version to be published and agreed to be accountable for all aspects of the work.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Katznelson L, Atkinson JL, Cook DM, Ezzat SZ, Hamrahian AH, Miller KK, American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of acromegaly—2011 update. Endocr Pract. 2011;17(Suppl 4):1-44.
doi pubmed - Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary. 2017;20(1):4-9.
doi pubmed - Broder MS, Chang E, Cherepanov D, Neary MP, Ludlam WH. Incidence and prevalence of acromegaly in the United States: a claims-based analysis. Endocr Pract. 2016;22(11):1327-1335.
doi pubmed - Burton T, Le Nestour E, Neary M, Ludlam WH. Incidence and prevalence of acromegaly in a large US health plan database. Pituitary. 2016;19(3):262-267.
doi pubmed - Dal J, Feldt-Rasmussen U, Andersen M, Kristensen LO, Laurberg P, Pedersen L, Dekkers OM, et al. Acromegaly incidence, prevalence, complications and long-term prognosis: a nationwide cohort study. Eur J Endocrinol. 2016;175(3):181-190.
doi pubmed - Rosendal C, Arlien-Soborg MC, Nielsen EH, Andersen MS, Feltoft CL, Kistorp C, Dekkers OM, et al. The changing landscape of acromegaly - an epidemiological perspective. Rev Endocr Metab Disord. 2024;25(4):691-705.
doi pubmed - Aagaard C, Christophersen AS, Finnerup S, Rosendal C, Gulisano HA, Ettrup KS, Vestergaard P, et al. The prevalence of acromegaly is higher than previously reported: Changes over a three-decade period. Clin Endocrinol (Oxf). 2022;97(6):773-782.
doi pubmed - Freda PU. Acromegaly: diagnostic challenges and individualized treatment. Expert Rev Endocrinol Metab. 2025;20(1):63-85.
doi pubmed - Esposito D, Olsson DS, Franzen S, Miftaraj M, Natman J, Gudbjornsdottir S, Johannsson G. Effect of diabetes on morbidity and mortality in patients with acromegaly. J Clin Endocrinol Metab. 2022;107(9):2483-2492.
doi pubmed - Wang K, Guo X, Yu S, Gao L, Wang Z, Zhu H, Xing B, et al. Patient-identified problems and influences associated with diagnostic delay of acromegaly: a nationwide cross-sectional study. Front Endocrinol (Lausanne). 2021;12:704496.
doi pubmed - Berkmann S, Brun J, Schuetz P, Christ E, Mariani L, Mueller B. Prevalence and outcome of comorbidities associated with acromegaly. Acta Neurochir (Wien). 2021;163(11):3171-3180.
doi pubmed - Daly AF, Jaffrain-Rea ML, Ciccarelli A, Valdes-Socin H, Rohmer V, Tamburrano G, Borson-Chazot C, et al. Clinical characterization of familial isolated pituitary adenomas. J Clin Endocrinol Metab. 2006;91(9):3316-3323.
doi pubmed - Horvath A, Stratakis CA. Clinical and molecular genetics of acromegaly: MEN1, Carney complex, McCune-Albright syndrome, familial acromegaly and genetic defects in sporadic tumors. Rev Endocr Metab Disord. 2008;9(1):1-11.
doi pubmed - Owens M, Stals K, Ellard S, Vaidya B. Germline mutations in the CDKN1B gene encoding p27 Kip1 are a rare cause of multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf). 2009;70(3):499-500.
doi pubmed - Daly AF, Tichomirowa MA, Beckers A. Update on familial pituitary tumors: from multiple endocrine neoplasia type 1 to familial isolated pituitary adenoma. Horm Res. 2009;71(Suppl 1):105-111.
doi pubmed - Malicka J, Swirska J, Nowakowski A. Familial acromegaly - case study of two sisters with acromegaly. Endokrynol Pol. 2011;62(6):554-557.
pubmed - Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414(6865):807-812.
doi pubmed - Arous C, Wehrle-Haller B. Role and impact of the extracellular matrix on integrin-mediated pancreatic beta-cell functions. Biol Cell. 2017;109(6):223-237.
doi pubmed - Slijepcevic D, Roscam Abbing RLP, Fuchs CD, Haazen LCM, Beuers U, Trauner M, Oude Elferink RPJ, et al. Na(+) -taurocholate cotransporting polypeptide inhibition has hepatoprotective effects in cholestasis in mice. Hepatology. 2018;68(3):1057-1069.
doi pubmed - Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98-107.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.